| AntiRetroScan 2.x | |
| (last updated January 10, 2009) | |
|
NOTICE This documentation refers to AntiRetroScan version 2.0, the currently running version. For documentation about AntiRetroScan 1.x, running from April 2001 to December 2008, see 1.x. |
|
| Changes with respect to AntiRetroScan 1.x | |
| AntiRetroScan version 2 was introduced in 2009 as a successor to the former versions 1.x developed at the University of Siena HIV Monitoring Service (SHMS) since April 2001. Version 2 still uses a mutation-drug scoring system as previous versions 1.x, an approach originally developed at Stanford University for HIVdb. However, the set of remodelling rules used in versions 1.x is not used any more. Instead, the score thresholds defining the five levels of predicted drug acitivity have been made drug-specific. In addition, drug class potency weighting factors have been introduced for computing a final regimen genotypic susceptibility score (GSS). | |
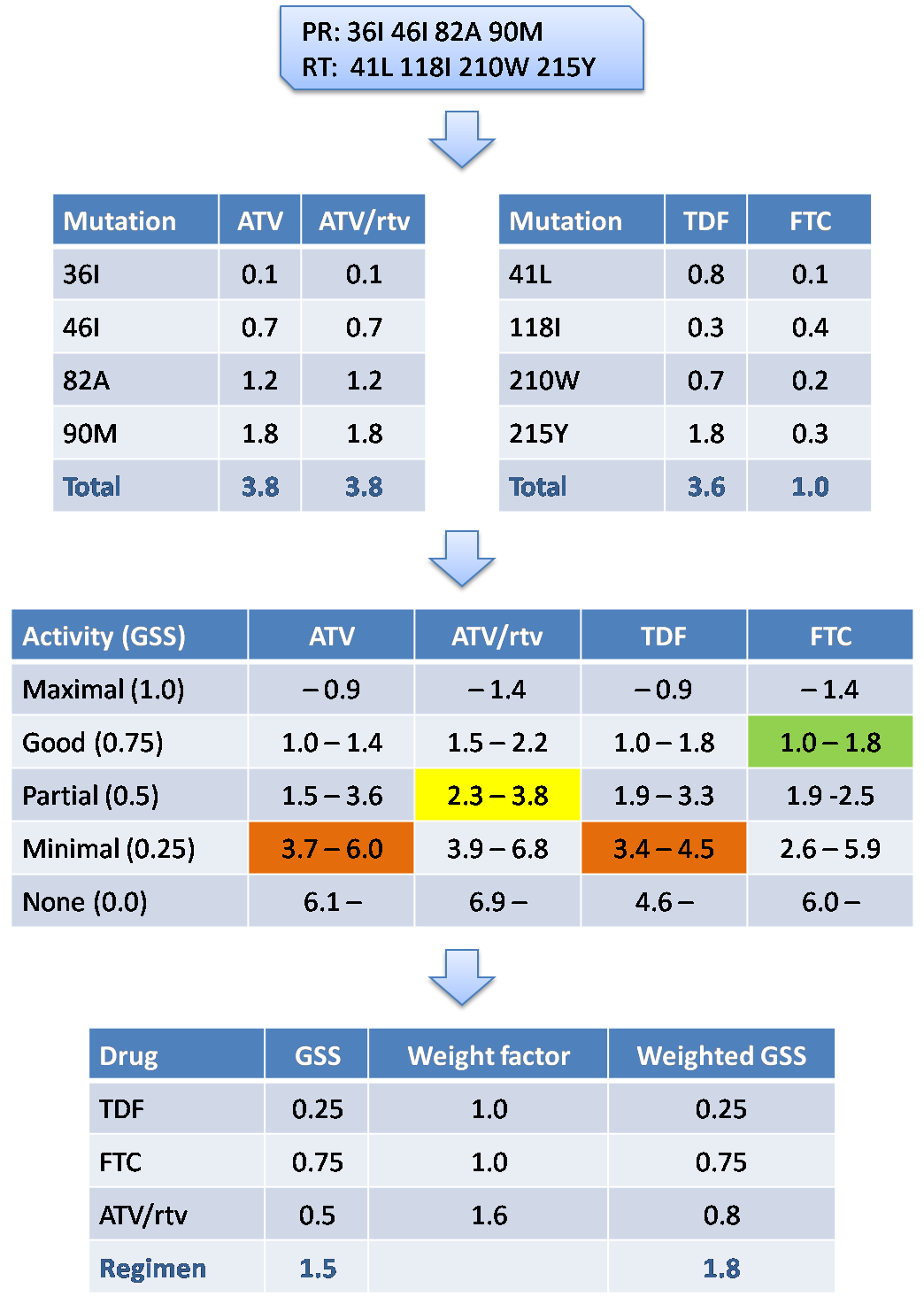
| How it works | |
| AntiRetroScan
2 analyzes the submitted sequence and extracts the mutations matching those
considered to be relevant for drug resistance in the
drug
resistance mutations reference
file. The same file has a score for each of these mutation for each antiretroviral
drug, based on the expected role of the mutation itself for that drug.
The sum of the scores obtained generates a total drug score and assigns each
drug to one of five activity categories defined by different ranges
of values in the drug activity range
reference file.
These ranges are also drug-specific and can be translated into percentage of
drug activity with respect to a 100% activity against the wild type virus.
Following a consolidated protocol, AntiRetroScan assigns a [0,1] normalized
genotypic sensitivity score (GSS) to the five activity categories as follows:
GSS(0%) = 0; GSS(25%) = 0.25; GSS(50%) = 0.50; GSS(75%) = 0.75; GSS(100%) = 1.0. Based on the expectation of a different potency with different drug classes, a drug class correction factor is finally applied to the GSS. Specifically, GSS for non-nucleoside reverse transcriptase inhibitors are multiplied by 1.2 and GSS for boosted protease inhibitors are multiplied by 1.6. To evaluate the overall potency of a specific combination regimen, use the sum of the GSS of the drugs included in the regimen. |
|
| Flowchart of AntiRetroScan | |
| AntiRetroScan output | |
| AntiRetroScan returns five predicted drug activity levels: | |
| 1. | |
| No resistance mutations found for the drug considered | |
| The drug is fully effective | |
| 2. | |
| The sequence contains only accessory mutations or mutations conferring resistance to other drugs but only minimal cross-resistance to the drug considered | |
| The response to the drug is highly likely | |
| 3. | |
| Resistance to the drug considered has started to develop or there are mutations conferring resistance to other drugs with partial cross-resistance to the drug considered | |
| The response to the drug is still possible but further development of resistance is likely if viral replication occurs | |
| 4. | |
| The resistance mutation pattern has a notable impact on susceptibility to the drug considered due to primary mutations selected by the drug itself or extensive cross-resistance caused by other drugs | |
| The response to the drug is very limited | |
| 5. | |
| There is complete resistance or cross-resistance to the drug considered | |
| The response to the drug is compromised, beneficial non-antiviral effects may persist | |
| Sources for definition of scores and rules | |
| The scores defined for each drug-mutation pair and the values are derived and periodically updated from the following sources: | |
| Correlation between genotype and in vitro susceptibility. These data are generated by in vitro analysis of laboratory or clinical HIV variants. This kind of information is normally derived from initial studies during the development of the drug and is the necessary starting point for further characterization of the resistance profile for a given drug. However, in vitro susceptibility assays are not devoid of shortcomings, e. g. it may be difficult to measure low resistance levels that are relevant in the clinic for some drugs or it may happen that mutations observed in vitro are extremely uncommon in vivo. | |
| Correlation between genotype and response to treatment in vivo. This information derives from clinical trials or observational studies and is presently considered the most important source for genotype interpretation algorithms. However, it is often difficult and prone to incorrect conclusions to obtain rules for individual drugs from analysis of data collected from combination therapy studies. Small-sized monotherapy pilot studies and ‘add-on’ studies, where a single drug is added to a background regimen, may provide useful data in this context. In recent clinical trials, new drugs have been evaluated when added to a genotype-guided background regimen and evaluated with respect to the optmized background regimen alone. | |
| Correlation between genotype and treatment history. This kind of analysis can be performed only on reasonably large databases. The association between treatment and variation in the HIV genome can be evaluated both in cross-sectional and longitudinal studies, provided that multiple sequences from the same patients at different time points are available. This approach may reveal novel associations and improve knowledge on the role of specific mutations. However, as antiretroviral treatment history is often complex, particularly in highly experienced patients, a variable proportion of mutations may have been selected by previous regimens but maintained by other drugs in the current regimen. | |
| Expected results | |
| AntiRetroScan 2.0 has been evaluated on a
large number of "Treatment change episodes" as a system to predict virological
response to antiretroviral regimens at 8 and 24 weeks following treatment
switch. When response was defined as a binary variable (success vs. failure) the
accuracy of AntiRetroScan was comparable to or higher than that of the commonly
used genotypic antiretroviral resistance interpretation systems (Stanford HIVdb,
ANRS, Rega). Main references: Zazzi M, Prosperi M, Vicenti I, Di Giambenedetto S, Callegaro A, Bruzzone B, Baldanti F, Gonnelli A, Boeri E, Paolini E, Rusconi S, Giacometti A, Maggiolo F, Menzo S, De Luca A on behalf of the ARCA Collaborative Group.Rules-Based HIV-1 Genotypic Resistance Interpretation Systems Predict 8-week and 24-week Virological Antiretroviral Treatment Outcome and Benefit from Drug Potency Weighting. 7th European HIV Drug Resistance workshop. 25-27 March 2009, Stockholm, Sweden. (pdf poster) Zazzi M, Prosperi M, Vicenti I, Di Giambenedetto S, Callegaro A, Bruzzone B, Baldanti F, Gonnelli A, Boeri E, Paolini E, Rusconi S, Giacometti A, Maggiolo F, Menzo S, De Luca A. Rules-based HIV-1 genotypic resistance interpretation systems predict 8-week and 24-week virological antiretroviral treatment outcome and benefit from drug potency weighting. J Antimicrob Chemother 2009;64:616-24. (MEDLINE) |
|
| Revisions | |
| - | 10/01/2009. Version 2.0. AntiRetroScan introduces drug-specific susceptibility levels and drug class-specific potency weights. |
| Prerequisites and limitation of the system | |
| AntiRetroScan accepts an HIV-1 protease
and/or reverse transcriptase sequence provided that at least protease
codons 10-93 and/or reverse transcriptase codons 41-219 are included.
If this relevant portion is incomplete the program warns and stops execution.
Protease and reverse transcriptase can be examined separately, however.
The reference sequence used for comparison with the submitted sequence is the subtype B consensus as available at the Los Alamos National Laboratory. The reverse transcriptase region beyond amino acid 400 is not considered. Warnings indicative of possible sequence quality problems include: |
|
| - | the presence of stop codons (TAA, TGA, TAG) |
| - | the presence of three-base degenerate codons (B, D, H, V), however considered as true by the system |
| - |
the presence of four-base degenerate codons (N), converted to the wild type base by the system |
| Subtype analysis is simply made by calculating the percentage of homology between the submitted sequence and the subtype and circulating recombinant form reference sequences as updated at the Los Alamos National Laboratory. Please note that while this approach is rapid and reasonably correct with ‘pure’ subtypes, recombinant forms should be subjected to formal phylogenetic analysis, if possible also including other regions of the same virus genome. | |
| Resistance to antiretroviral drugs is a rapid evolving field, particularly with most recent compounds. The necessary updates of the system thus imply that the same genotype may be interpreted differently in different time periods (i. e. algorithm versions). | |
| Contact us | |
| Address your questions or comments to Maurizio Zazzi. | |
{kind=link}